
Articles
Vol. 36 No. 3 (2014)
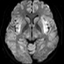
Hyperammonemic coma in a patient with late-onset OTC deficiency
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Received: 13 November 2014
Accepted: 13 November 2014
Accepted: 13 November 2014
2195
Views
3012
Downloads